Description
August 19, 2019The permutation test is an often used test procedure for determining statistical significance in brain network studies. Unfortunately, generating every possible permutation for large-scale brain imaging datasets such as HCP and ADNI with hundreds of networks is not practical. We present the novel transposition test that exploits the underlying algebraic structure of the permutation group in drastically speeding up the permutation test. If you are using either data or code below, please reference Chung et al. (2019), where the method is first published. The transposition can be further used in the rapid computation of twin correlations, which is traditionally computed using the intra-class correlations.
The package will show how to perform the transposition test, which generates 100-1000 (depending on the sample sizes) more permutations than the standard permutation test in the same computational run time. The complete package is zipped into matlab.zip. The code SIMULATION.m will duplicate the simulation studies given in [1]. Due to random nature simulation, the results vary. However, the codes can handle vector data. For more complex data such as images and graphs/networks, you need to vectorize the data first. The power point presentation of the method is given here.
1) Permutation test
August 19, 2019; December 5, 2021For data vectors x and y, we define the test statistic as a function using the built-in inline.m function:
stat = inline('max(abs(mean(x)-mean(y)))').
This is the test statistic used in the simulation study of common 1-cycle basis in study [2]. For two-sample t-stat used in study [1], we define
stat = inline('mean(x)-mean(y)).*sqrt(m*n*(m+n-2)./((m+n)*((m-1)*var(x)+(n-1)*var(y))))');
The standard permutation test is then performed by running
[stat_s, time_s] = test_permute(x,y,per_s, stat)
where per_s is the number of permutations. The function inputs function as an augment. The resulting statistical result is stored in stat_s. The run time of the test is stored in time_s.
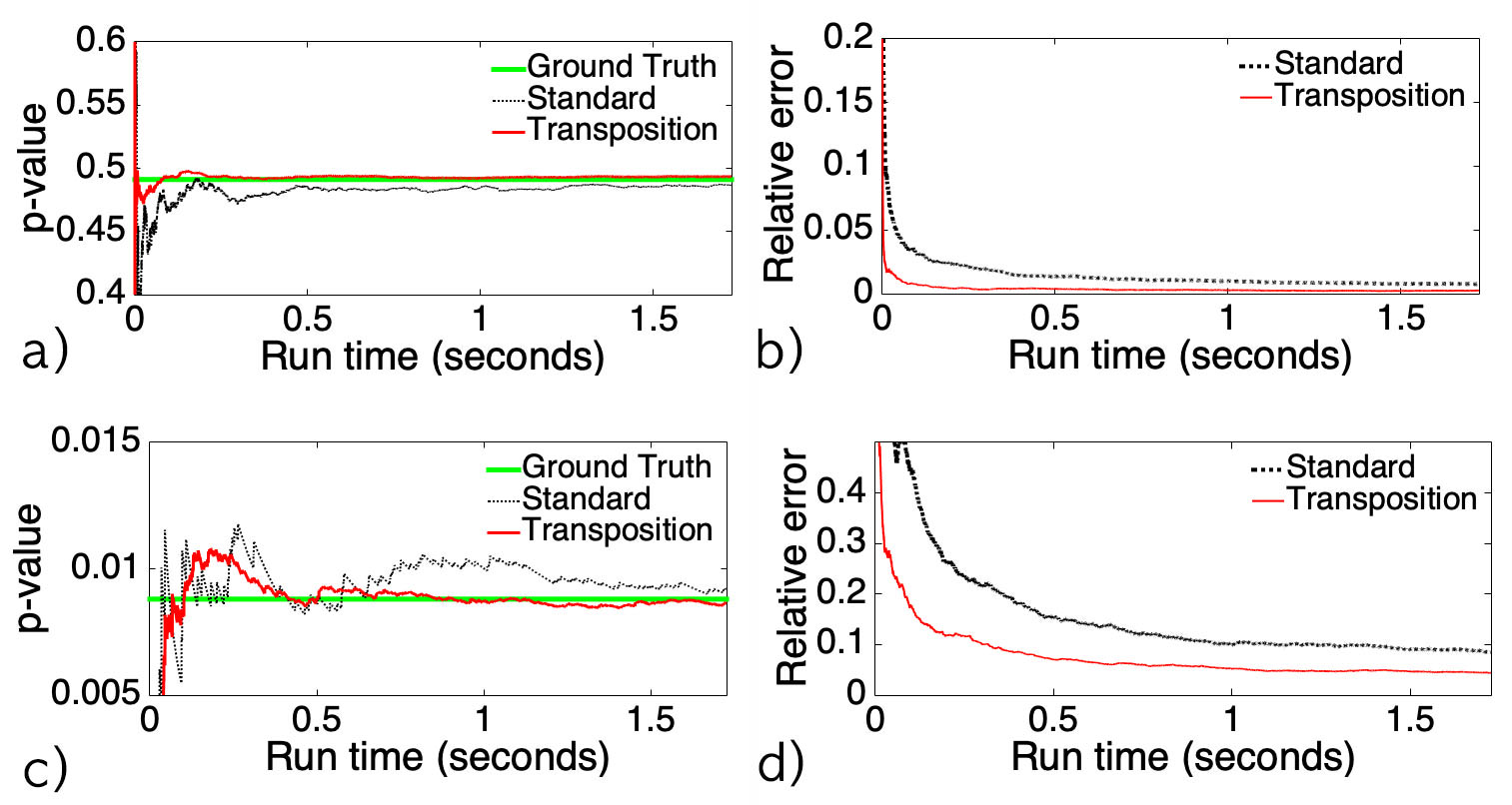
Figure 1. Simulation result showing superior performance of the transposition test over the standard permutation test. Read Chung et al. (2019) for details.
2) Transposition test
August 19, 2019The transposition test is done by running
[stat_t, time_t] = test_transpose(x,y,per_t, stat)
where and per_t is the number of permutations. The resulting two-sample t-stat result is stored in stat_t. The run time of the test is stored in time_t. test_transpose.m starts the sequence of random transpositions from the permutation of [x y].
Accelerating the convergence of the transposition test
August 19, 2019Chung et al. (2019) explained how to accelerate the convergence and avoids possible bias by interjecting the random permutations within the sequence of transpositions. That can be easily achieved by running iterations
stat_t=[];
for i=1:1000
[stat, time] = test_transpose(x,y,per_t/1000);
stat_t=[stat_t; stat];
time_t=time_t+time
end
where the sequence of transpositions of length per_t are broken into 1000 independent sequences. The number of sequences needs to be determined empirically but the length of each sequence should be around 100-1000.
Transposition test on the whole brain or network
August 19, 2019The transposition test on the whole brain or network can be done by vecotrizing data. Suppose we generated the following simulated vector data, where group 1 has m=10 subjects and group 2 has n=12 subjects over l=1000 voxels.
m=10; n=12; l=1000; per_t=100000;
x= normrnd(0,1,m,l);
y= normrnd(0.1,1,n,l);
Then the transposition test is done as
[stat_t, time_t] = test_transpose(x,y,per_t);
The p-value is then computed as
observed=(mean(x)-mean(y)).*sqrt(m*n*(m+n-2)./((m+n)*((m-1)*var(x)+(n-1)*var(y))))
pvalue_t = online_pvalue(stat_t, observed);
3) Twin correlation computation using
transpositions
June 03, 2020
The above transposition method can be further used
to rapidly compute the twin correlations, which can
be a serious computational bottleneck for large
sample sizes. The method is illustrated with a
simple example. Consider 5 twins, where each twin is
paired along the rows.
-2.2439 -0.0720
1.3997 -0.2961
-0.0870 2.6186
1.3410 -1.0642]
x=a(:,1)
y=a(:,2)
The twin correlation based on 5000 transpositions is given by running
per_t=5000
[stat_t, time_t] = transpose_corr(x,y,per_t)
where time_t is the run time and stat_t is the average correlation of all the correlations over transpositions. It is sequentially computed in the online fashion to save computer memory. Note the length of stat_t is the number of transpositions. per_t. stat(5000) is the average correlation over 5000 transpositions. Since the result depends on random transpositions, the results randomly change. Thus, it is necessary to average the results. The number 5000 is empirically chosen from the plot (Figure 2). Empirically we choose per_t such that the estimated the correlation values does not change in 2 decimal places,
In this example, we performed the simulation 10 times and the results are averaged. The standard deviation of 10 simulation should be smaller 0.01. This grantee the convergence within two decimal places in average.
figure
corr=[];
for i=1:10
per_t=5000;
[stat_t, time_t] = transpose_corr(x,y,per_t);
hold on; plot(stat_t);
corr=[corr stat_t(per_t)];
end
[mean(corr) std(corr)]
-0.1464 0.0043

Figure 2. Correlation estimation using 5000 random transpositions. 5000 is chosen since there is no more fluctuations and it is a large enough round number. The result needs to be averaged to get more robust estimate.
In the above code, stat_t should converge to the true underlying twin correlation as per_t increases. transpose_corr.m can input matrix data, where x and y can be the input data of size m (number of subjects) by p (number of variables/connections/voxels per subject). For instance, it will input 5x3 data matrices:
x=0.1 +
normrnd(0,1,5,3);
y= normrnd(0,1,5,3);
per_t=5000;
[stat_t, time_t] = transpose_corr(x,y,per_t);
Heritability and its p-value computation using transpositions
June 11, 2020
Given the following data, heritability index (HI)
is computed as follows. Assume there are 50 MZ-twins
(xM and yM paired) and 40 DZ-twins (xD and yD
paired):
xM=0.1 + normrnd(0,1,50,1);
yM= xM + normrnd(0,0.1,50,1);
xD=0.1 + normrnd(0,1,40,1);
yD= xD + normrnd(0,1,40,1);
Based on transpose_corr.m,
heritability index (HI) is computed as follows
per_t=10000;
[stat_MZ, time_t] = transpose_corr(xM,yM,per_t);
[stat_DZ, time_t] = transpose_corr(xD,yD,per_t);
HI = 2*(corr_MZ-corr_DZ)
which gives HI value of 0.8537 indicating strong
heritability. This number will randomly
change. In this small sample example,
per_t=10000 gives sufficient convergence. HI-value
0.3833 is our observation. We will determine the
statistical significance of 0.3833 using the
empirical distribution of HI.
per_s=100000;
[all_t, time_t] = transpose_heritability(xM,yM,xD,yD,
per_s);
where per_s is the total number of transpositions
between MZ- and DZ-twins. 50% of transpositions are
within twins. The optimal ratio will be later
determined. all_t outputs the every possible
HI-values over transpositions within and between
twins. It is implemented following the formula
sheet: transpose-covariance.pdf.
per_s has to be extremely large for convergence. For
HCP data, it is expected per_s is no less than 10
million for convergence. But this has to be
determined empirically that gives sufficient
convergence. One visual trick to determine if
100000 is big enough is by checking if the empirical
probability distribution is smooth enough (Figure
3):
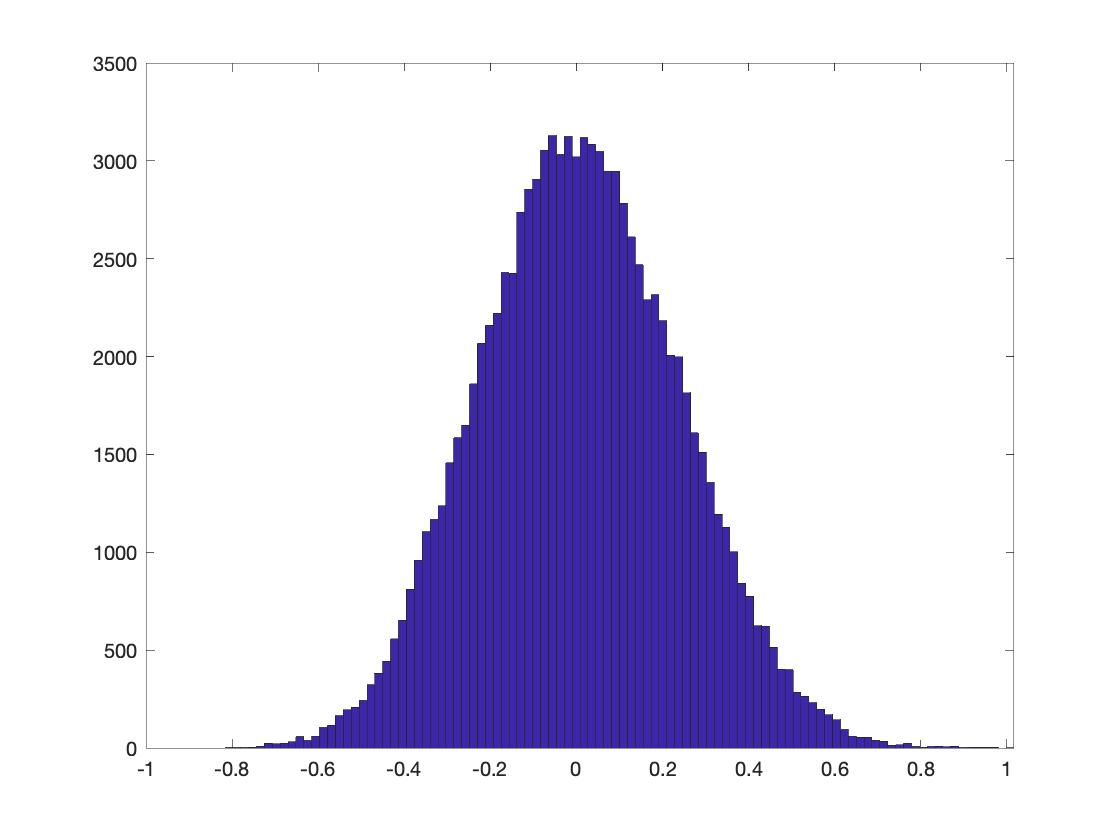
Figure 3. The histogram (unormalized empirical distribution) of all_t.
Then p-value is given by
pvalue = online_pvalue(all_t, HI)
figure; plot(pvalue)
where pvalue(1000) is the p=value computed using up to 1000 transpositions. pvalue(end) is the p-value using all the 100000 transpositions.
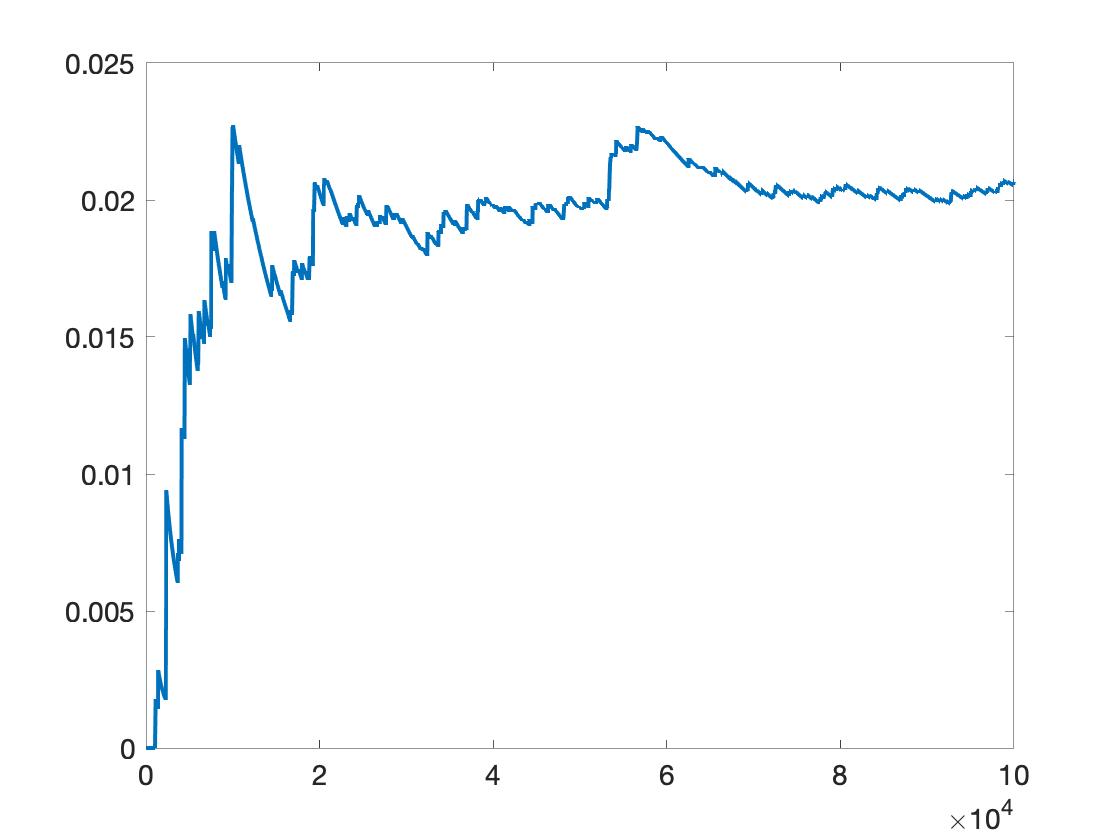
Figure 4. p-value plot of all_t over the number of transpositions. It shows, per_s=100000 is not enough. Likely 10 times more transpositions are needed.
Here, we have shown how to compute uncorrected
p-value for one variable/connection per subject. It
should not be hard to extend it and compute
the corrected p-value for multiple
variables/connections per subject.
Reference
[1] Chung, M.K., Xie, L, Huang, S.-G., Wang, Y., Yan, J., Shen, L. 2019 Rapid acceleration of the permutation test via transpositions, International Workshop on Connectomics in NeuroImaging, Lecture Notes in Computer Science (LNCS) 11848:42-53. An earlier version with different application arXiv:1812.06696
[2] Anand, D.V., Dakurah, S., Wang, B., Chung, M.K. 2021 Hodge-Laplacian of brain networks and its application to modeling cycles. arXiv:2110.14599
