The R package "WGDgc" uses gene count data (number of gene copies) across multiple gene families and multiple species. The background process of gene duplications and gene losses is modelled by a birth and death process. In addition, whole genome duplications (WGD) and whole genome triplications (WGT) are modelled with an instantaneous duplication (or triplication) of all genes present at the time of event, with a probability of retaining the extra gene (or of retaining one of both extra copies for whole genome triplications).
The duplication (birth) rate, loss (death) rate and retention rate at each WGD or WGT event are estimated simultaneously using maximum likelihood. A shifted geometric prior distribution is used for the unobserved number of genes at the root. The likelihood can be conditioned on the data collection process. This method is fast. It is further explained here:
Charles-Elie Rabier, Tram Ta and Cécile Ané (2014). Detecting and Locating Whole Genome Duplications on a phylogeny: a probabilistic approach. Molecular Biology and Evolution, 31(3):750-762, abstract.
WGDgc depends on the R packages ape, phylobase and phyext.
You can install ape and phylobase from CRAN using the R command
install.packages(c("ape","phylobase")) .
The phyext package is no longer on CRAN, but you can install it from source like this:
install.packages("https://cran.r-project.org/src/contrib/Archive/phyext/phyext_0.0.1.tar.gz", repos = NULL, type="source")install.packages("https://github.com/cecileane/WGDgc/archive/v1.3.tar.gz", repos=NULL, type="source")phytools
after loading phyext.
phytools is not needed
for WGDgc, but its function read.simmap will
interfere with the function read.simmap in phyext.
This method aims to detect whole genome duplications, but is much slower than the method based on gene count data above (see Rabier et al. 2014 above). SPIMAPWGD can also be used to improve gene tree estimation when whole genome duplications have occurred in the group of interest. It is a version of the software SPIMAP of Matthew Rasmussen and Manolis Kellis, which incorporates Whole Genome Detection (WGD) events.
For background on SPIMAP, see "A Bayesian approach for fast and accurate gene tree reconstruction", Rasmussen and Kellis, Mol. Biol. Evol. 28(1):273-290.2011, and documentation here. SPIMAPWGD uses the same arguments as SPIMAP and also new arguments. Here, you will find some explanations on how to run SPIMAPWGD. Some examples are given at the end of the page. A version of SPIMAPWGD is available on github.
Note that SPIMAPWGD will generate the following files : *.tree (final gene tree), *.duplosslastree (total losses, losses at WGD, total duplications, duplications at WGD for final gene tree), *.completeloglikelihood (approximated loglikelihood), and *.topologyprob (topology prior).
SPIMAPWGD requires a species tree in the Newick format. In order to include a Whole Genome Duplication in the species tree, just add a supplementary node called "WGDp" where p is the value of the probability of loss at the WGD.
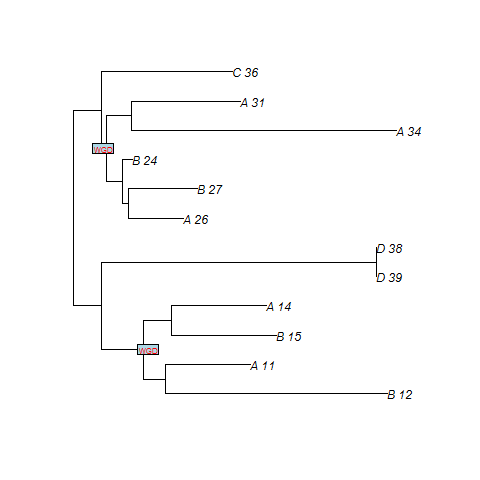
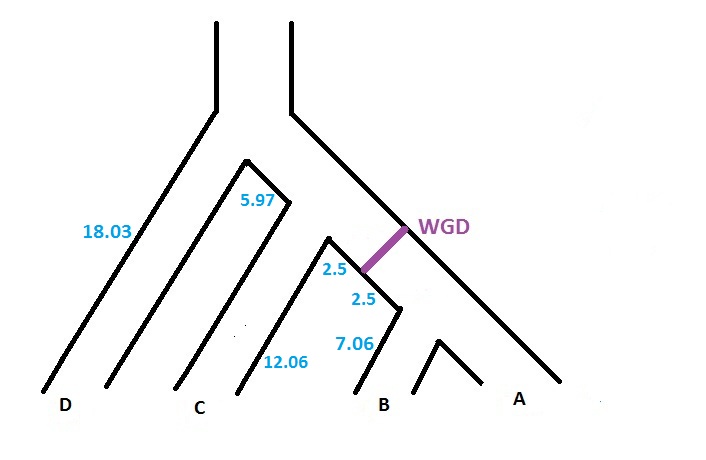
For instance, if we consider a species tree with 4 taxon (A,B,C,D), and if we want to specify that a WGD, with probability of loss 0.10,
is present just at the middle of the branch located before the speciation A|B, the input file corresponding to the figure below will be
((((A:7.06,B:7.06):2.50)WGD0.10:2.50,C:12.06):5.97,D:18.03);
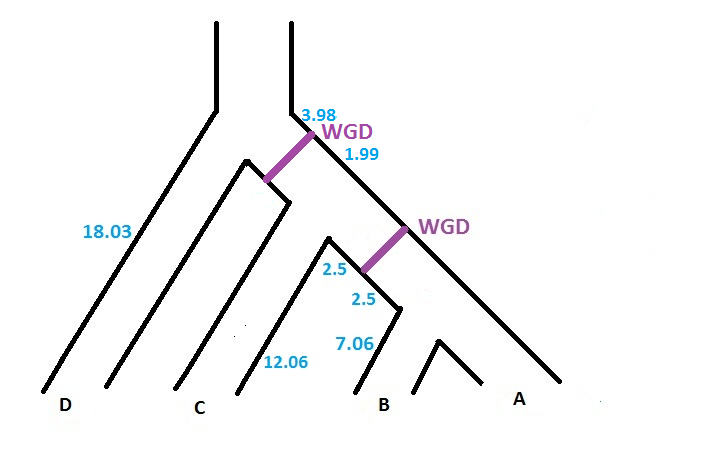
The software can handle several WGDs, each with its own loss probability. However, each WGD has to be located on a different branch of the species tree. So, if we want to add another WGD (with loss probability 0.12) on the branch located just above the previous WGD (cf. figure below), the input file will be
(((((A:7.06,B:7.06):2.50)WGD0.10:2.50,C:12.06):1.99)WGD0.12:3.98,D:18.03);
We just have to use the same input file as in SPIMAP. In other words, we do not have to take into account that some WGD are present inside the species tree
As in the original version of SPIMAP, the software gives the reconciliation (with the option -r). It now specifies which nodes are reconciled at the WGD. Note that when several WGDs are located in the species tree, before interpreting the reconciliation file (.recon), we have to know the code associated to each WGD. This code is given on the screen when we run SPIMAPWGD. In particular, SPIMAPWGD describes the 2 WGDs of the figure above in the following way :
WGD event number0:
child node : n5
loss probability: 0.100000
percent distance: 0.500000
total distance: 5.000000
distance 'before': 2.500000
distance 'after': 2.500000
WGD event number1:
child node : n4
loss probability: 0.120000
percent distance: 0.666667
total distance: 5.970000
distance 'before': 3.980000
distance 'after': 1.990000
'Total distance' gives the length of the branch on which the WGD is placed.
'Percent distance' indicates where the WGD is located on the branch, as a percentage of the total distance starting from
the parent node.
Distances 'before'/'after' are the distances before/after the WGD.
We can see that the oldest WGD has the number 1, whereas the most recent has the number 0.
Recall that the reconciliation file (.recon) has 3 columns. The first column refers to the gene tree whereas the second column refers to the species tree. On each row, a node of the gene tree (first column) is reconciled to a node of the species tree (second column). The third column indicates the corresponding evolutionary event : 'gene', 'speciation', 'dup' (for duplication), 'WGD'. Note that when the evolutionary event is a WGD, column 2 also gives the number associated the WGD.
We give here an example of gene tree (simulated from our species tree with only one WGD) obtained by SPIMAPWGD, and also some rows of the reconciliation file (.recon) :
| A_11 | A | gene |
| B_12 | B | gene |
| B_15 | B | gene |
| C_36 | C | gene |
| n4 | n5 | spec |
| n6 | D | dup |
| n8 | WGD_at0 | WGD |
| n3 | WGD_at0 | WGD |
| ... | ... | ... |
SPIMAPWGD finds two duplications at the WGD. n8 and n3 are the nodes of the gene tree that map at the WGD (cf. figure). Note that file .duplosslasttree gives the total number of losses, the number of losses at the WGD, the total number of duplications and the number of duplications at the WGD.
| A_10 | A | gene |
| B_11 | B | gene |
| C_12 | C | gene |
| n3 | WGD_at0 | WGD |
| n2 | WGD_at1 | WGD |
| n8 | WGD_at0 | WGD |
| ... | ... | ... |
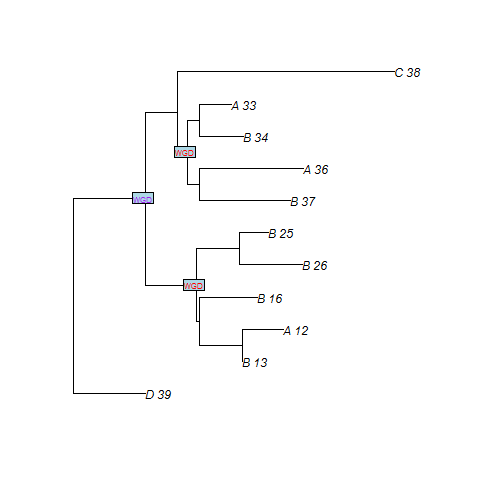
SPIMAPWGD finds three duplications at the WGDs. n2 is the node of the gene tree which maps to the WGD number 1. n3 and n8 are the nodes which maps to the WGD number 0. We recall that WGD number 1 is the oldest WGD whereas WGD number 0 is the most recent.
For more details, download TUTORIAL.tar.gz and open the file README.sh. You will find the same examples as the ones present on this page. In the folder "dnawith2WGD" (resp. "dnawith2WGD"), you will find 20 data sets, generated with the species tree with one WGD (resp. two WGDs).
As explained in the paper, we ran SPIMAPWGD on a data set of 16 fungal species. We considered the same gene clusters as in Butler et al. (Nature 2009), that is to say 9209 gene families, downloaded from http://compbio.mit.edu/candida . The fungal species tree comes also from this recent study. The WGD was placed at the middle of the branch (arbitrarily) of the species tree where a putative WGD happened in yeast. An example of species tree with probability of loss 0.4 can be downloaded here. You will also find a table which summarizes our results, and the gene trees found for each family (gene family sizes: 3 to 40, 41 to 56, 57 to 130 , readme.txt).